Cigarette smoke-induced COPD is a debilitating disorder of the lung. It is the 3rd leading cause of death worldwide, and its prevalence is increasing. Nearly as many will die of COPD this year as AIDS and cancer combined. The cost of the COPD problem last year alone was ~$23 trillion. COPD is characterized by chronic airway inflammation, mucus hypersecretion, airway remodeling, and emphysema that lead to reduced lung function and breathlessness. Systemic effects also have been observed in the skeletal muscle, heart, and gastrointestinal tract of those who smoke. Moreover, COPD patients are more susceptible to respiratory infections. Because the mechanisms that lead to COPD and its sequelae are poorly understood at the molecular level, there are no effective treatments that hinder disease progression or its reversibility.
A major factor that hampered the study of COPD was the lack of a small animal model that recapitulates the hallmark features of the disease in a more reasonable time frame. To that end, my collaborators and I recently developed a mouse model of COPD in which we delivered tightly controlled amounts of cigarette smoke directly into the airways. The treated mice exhibited the major characteristic features of COPD observed in humans after only 8 weeks of smoke exposure, thereby facilitating the discovery and/or testing of the efficacy of new therapeutics. The model also enabled us to elucidate the cellular, biochemical, and molecular mechanisms that underpin the pathogenesis of COPD.
Although MCs have been casually linked to the pathogenesis of COPD in humans, the relevant factors exocytosed from these immune cells have not been identified. MC numbers are increased in inflammatory infiltrates in COPD patients, and their presence is associated with reduced lung function, airway remodeling, and emphysema. The levels of hTryptase-β in the sputum correlated with the severity of COPD in one study, and the exposure of IL-3-dependent mouse MCs to cigarette smoke-treated culture medium resulted in increased expression of mMCP‑6. As noted in the bacterial infection and arthritis studies, mMCP-6 promotes inflammation, chemokine expression, and macrophage and neutrophil chemotaxis which are all hallmark features of COPD. Like human MCs, mouse MCs express three tryptases. Using the novel transgenic mouse strains we created, we discovered that all three MC-restricted tryptases had significant adverse roles in our COPD.animal model due to their overlapping substrate specificities [Beckett-Stevens et al., J. Allergy Clin. Med. 2013;131:752) (view Beckett-Stevens et al.) and Hansbro-Hamilton et al., J. Biol. Chem. 2014;289:18214) (view Hansbro-Hamilton et al.)] (see Figure 8).
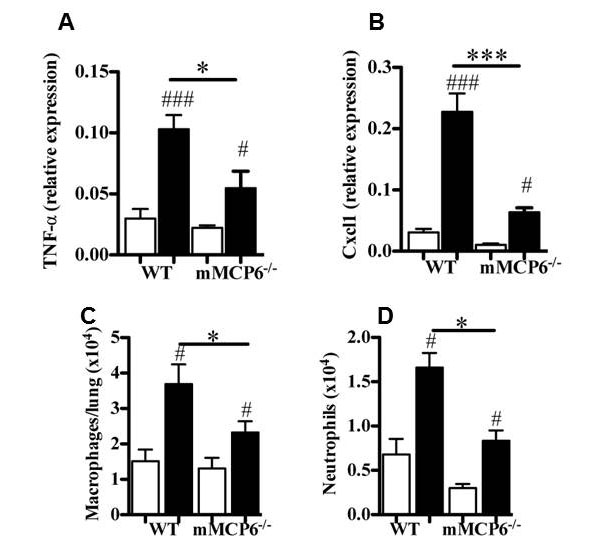
Fig. 8. Reduced COPD in transgenic mice lacking the tetramer-tryptase mMCP-6. Exposure of mMCP-6-null mice to cigarette smoke results in reduced expression of proinflammatory cytokines (A) and chemokines (B) and reduced numbers of macrophages (C) and neutrophils (D) in the lung relative to smoke-treated WT mice (from Beckett-Stevens et al., J. Allergy Clin. Med. 2013;131:752).
Humans who have inactivating mutations in the gene that encodes the serpin A1AT are at increased risk of developing emphysema even if they never smoked, thereby documenting the importance of a A1AT/tryptic protease balance in the lungs. We discovered that hPRSS31 is rapidly inactivated by A1AT (Wong et al., J. Biol. Chem. 2002;277:41906) (view Wong et al.). The fact that the genetic loss of a natural inhibitor of this MC‑restricted tryptase dramatically increases the risk of lung inflammation in humans supports our experimental mPrss31-COPD data. Based on our mouse findings, the therapeutic potential of MC tryptase inhibitors may only be realized when both hTryptase-β and hPRSS31 are inactivated.
Comments are closed.